
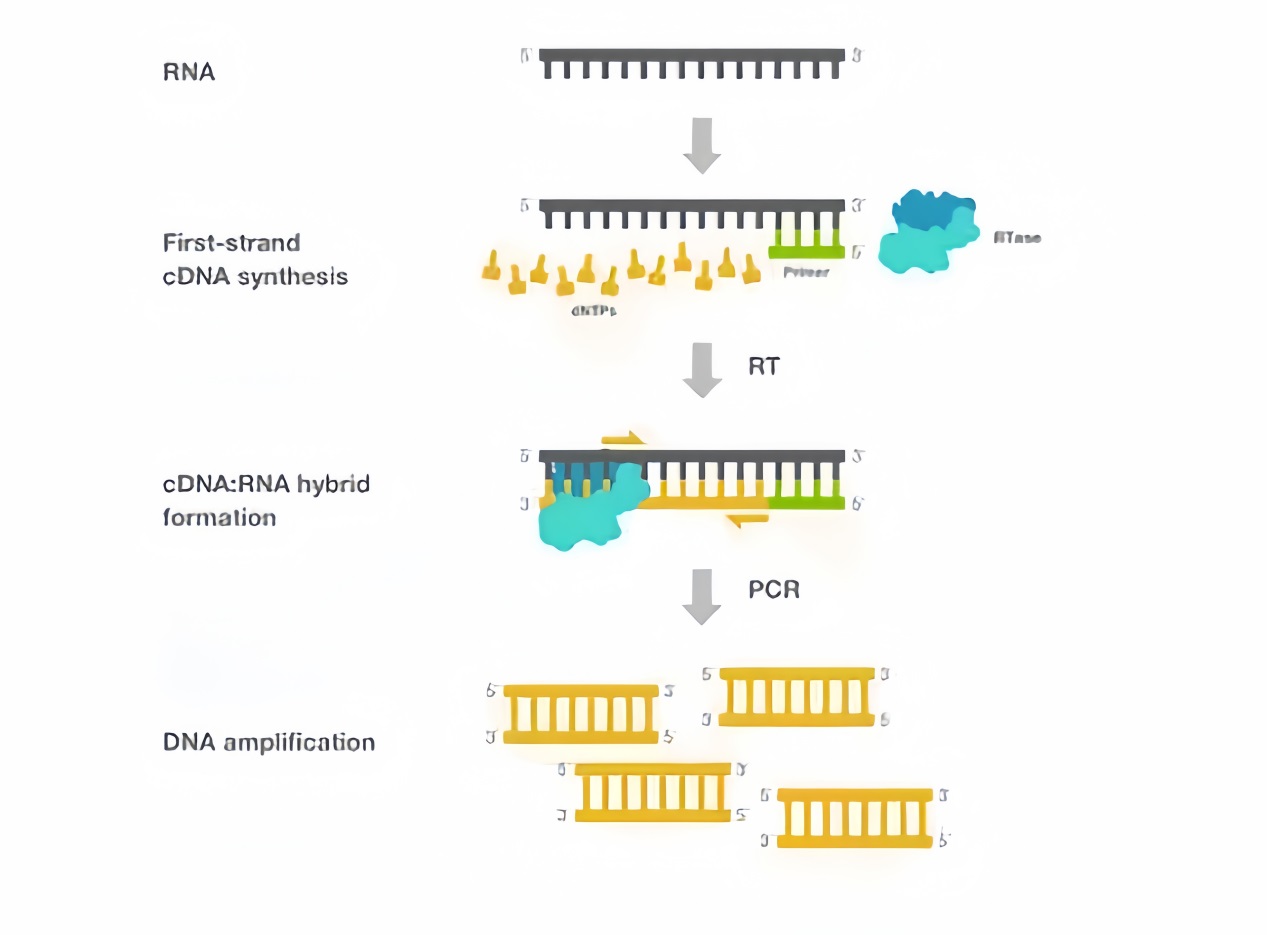
Primers refer to large molecules with specific nucleotide sequences that are stimulated and synthesized at the beginning of nucleotide polymerization, and are connected to reactants in the form of hydrogen bonds. Such molecules are called primers. Primers are usually two artificially synthesized oligonucleotide sequences. One primer complements a DNA template chain at one end of the target region, while the other primer complements another DNA template chain at the other end of the target region. Its function is to serve as the starting point for nucleotide polymerization, and nucleic acid polymerase can start synthesizing new nucleic acid chains from its 3 ‘end. Primers artificially designed in vitro are widely used in polymerase chain reaction, sequencing, and probe synthesis.
DNA polymerase can only add new deoxyribonucleotides to the existing 3 ‘end during PCR amplification, forming new phosphodiester bonds with the phosphate groups of the new nucleotides. The function of primers is to bind to the sense chain during PCR annealing, thereby providing a 3 ‘- terminal hydroxyl group.
The purpose of PCR primer design is to find a suitable pair of nucleotide fragments that can effectively amplify template DNA sequences. As mentioned earlier, the quality of primers directly affects the specificity and success of PCR. It is impossible to have a comprehensive rule for primer design to ensure the success of PCR, but following certain principles can help with primer design.
There are three basic principles for primer design: firstly, the sequences of primers and templates should be closely complementary. Secondly, stable dimers or hairpin structures should be avoided between primers. Thirdly, primers should not trigger DNA polymerization reactions (i.e. mismatches) at non target sites on the template. The specific implementation of these three basic principles requires considering many factors, such as primer length, product length, sequence Tm value (melting temperature), internal stability of primer template forming double chains (reflected with ∆ G value), energy value of primer dimer and hairpin structure formation, The triggering efficiency at false priming sites, the GC composition of primers and products, and so on. If necessary, primers need to be modified, such as adding restriction endonuclease sites, introducing mutations, etc.
Principles of primer design
- Basic principle
1.1. Primer length: 15-30bp, commonly around 21bp.
1.2. The appropriate span for bow | object amplification is 200-500bp (the specific length is designed according to the experimental purpose).
1.3. The optimal G+C content should be between 40% -60%. If G+C is too small, the amplification effect is not good, and too many are prone to non-specific bands. It is best to randomly distribute ATGC to avoid a string arrangement of more than 5 purine or pyrimidine nucleotides.
1.4. Avoid secondary structures within the bow | object, and avoid complementation between two primers, especially at the 3 ‘end, otherwise it will form primer dimers and produce non-specific amplification bands
1.5. The base at the 3 ‘end of the primer should be strictly paired, especially the second to last base, to avoid PCR failure due to non pairing of the end base
1.6. Appropriate restriction sites are present or can be added to primers, and it is best for the amplified target sequence to have suitable restriction sites, which is beneficial for restriction analysis or molecular cloning
1.7. The specificity of the primers should be such that they have no significant homology with other sequences in the nucleic acid sequence database.
1.8. Primer quantity: Each primer has a concentration of 0.1-0.5 μ It is better to produce the desired result with the lowest amount of bow material. A high concentration of bow material can cause mismatches and non-specific amplification, and can increase the chances of bow material forming aggregates. - Annealing temperature and time
The annealing temperature and annealing time depend on the length of the target molecule, base composition and concentration, and the sequence length of the target gene - The refolding temperature of the primer Tm=4 (G+C)+2 (A+T)
3.1. Refolding temperature=Tm value – (5-109c)
3.2. Within the allowable range of TM values, selecting a higher refolding temperature can reduce the non-specific binding refolding time between the arch and template, which is generally 30-60 seconds. The specific time depends on the length of the amplification product sequence.
Modifying primers/probes
Modifying primers/probes refer to the addition of chemical groups or changes in the base and phosphodiester bond on the sequence of regular primers to achieve target detection, prevent degradation, increase stability, and other purposes. There are more than 300 types of modifications, which can be divided into 3 ‘- end modification, 5’ – end modification, and intermediate modification according to their positions. Fluorescent labeled modifying primers are usually referred to as probes.
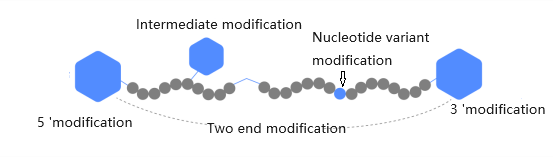
Common types of partial modifications
- Biotin
Biotin modified oligonucleotides can tightly bind to streptavidin proteins, which can be labeled with fluorescent dyes and enzymes or attached to solid biological surfaces as intermediate ligands. Different molecular biology and purification methods include biotin modification. Biotin modification can utilize C6 or TEG arms to be added at the 5 ‘or end of oligonucleotides. Biotin TEG needs to be purified, and intermediate biotin modification can be added through dT bases. This form requires more purification steps. Primer biotin labeling can be used for non radioactive immunoassay to detect proteins, intracellular chemical staining, cell separation, nucleic acid separation, hybridization detection of specific DNA/RNA sequences, ion channel conformational changes, etc. - Digoxin
Digoxin is a steroid substance isolated from the plant Rehmannia glutinosa, as the flowers and leaves of the plant are the only natural source of this substance, anti digoxin antibodies do not bind to other biological substances.
Digoxin is connected to the C5 position of uracil via an 11 atom intermediate arm. Hybridized digoxin probes can be detected by anti digoxin antibodies, which are typically conjugated with alkaline phosphatase, peroxidase, fluorescein, or colloidal gold. Or there may be no occasional anti digoxin antibodies but occasional antibodies. The probe labeled with digoxin can be used for various hybridization reactions, such as DNA-DNA hybridization (Southern blotting), DNA-RNA hybridization (Northern blotting), dot blotting, clone hybridization, in situ hybridization, and enzyme-linked immunosorbent assay (ELISA). - Phosphorylation
5 ‘phosphorylation is achieved by adding B-cyanoethyl chemical reaction to the sugar ring at the 5’ end of the primer, rather than the last base; 3 ‘phosphate is connected to the solid support medium, so in the first cycle of synthesis, the base is coupled with it. The 3 ‘phosphorylation modification has the function of preventing the extension of polymerase. 5 ‘phosphorylation can be used for splicing, cloning, gene construction, and ligase catalyzed linkage reactions. Phosphorylation is also used in experiments related to resistance to exonuclease digestion to prevent DNA polymerase catalyzed DNA strand elongation reactions. - Phosphorothioate
As the number of thio bases increases, the Tm value of oligonucleotides decreases. To reduce this effect, 2-5 bases at both ends of the primer can be thio modified. Typically, 3 bases each, 5 ‘and 3’, can be selected for thio modification. Thio modified oligonucleotides are mainly used in antisense experiments to prevent degradation by nucleases. - Internal amino modification
Mainly using C6-dT aminolinker to add to thymine residues for internal modification. The modified amino group is 10 atoms away from the main chain and can be used for further labeling and enzyme linkage (such as alkaline phosphatase). - 5 ‘amino modification
It is mainly divided into two types: 5’C6 amino modification and 5’C12 amino modification. The former can be used to connect compounds that do not affect their function even when they are close to oligonucleotides, while the latter is used for affinity purification of functional groups and some fluorescence labeling, especially when fluorescence may be quenched due to labeling too close to the DNA strand. It can be used to prepare functionalized oligonucleotides and is widely used in DNA microarrays and multi label diagnostic systems. Currently, it is mainly used to connect compounds that do not affect their function even if they are close to oligonucleotides. - 3 ‘amino modification
Mainly C6 amino modification, it can be used to design new diagnostic probes and antisense nucleotides, such as the 5 ‘end can be labeled with highly sensitive 32P or fluorescein, while the 3’ end can be amino modified for other connections. In addition, 3 ‘modification can inhibit the enzymatic hydrolysis of 3’ exonuclease, making it suitable for antisense experiments.
Source:
https://baike.baidu.com/
https://zhuanlan.zhihu.com/p/567004409
https://zhuanlan.zhihu.com/p/133676612

{kind=link}